You have more questions about SWATH? Maybe this will help!
We know it. Mass spectrometry is quite complicated. OK, it is very complicated! The different concepts behind the technology is uses and/or the acquisition mode it offers are pretty hard to understand. This is why we chose to write this post to answer some of the questions that we have been asked about SWATH. This post is directly related to our blog post on the SWATH acquisition mode, so make sure you read it before digging into this one!

Question 1 : How is the ion library for a SWATH analysis generated? I read in the application note that it was by IDA.. but is there another name for this acquisition method? It’s not the same as shotgun proteomics or MRM, right?
Answer : The database is indeed generated by the data dependant acquisition (DDA), that may also be called information dependant acquisition (IDA). This acquisition mode is used to do shotgun proteomics, but it’s not the same as the MRM mode. The MRM mode is used for targeted quantification of a molecule or peptide and has no use in the generation of an ion library.
Now, in every mass spectrometry run, the machine follows the instructions that are listed in the method that we want to use. Since the machine is very quick, it can perform these instructions in a very short time. It takes the machine only 1.6 seconds to complete all the instructions in our method! And as soon as the machine is done with the instructions, it starts another cycle. This means that the machine records what's in the sample every 1.6 seconds for as long as the HPLC gradient lasts.
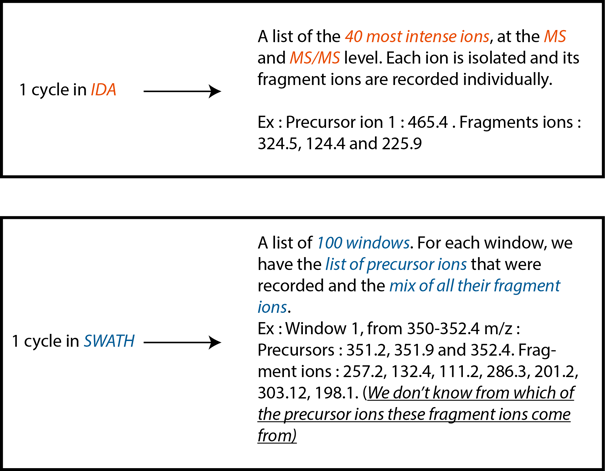
Knowing this, we can now explain how the IDA method works. To generate an ion library, the machine makes a survey scan (for something like 0.25 sec) of all the ions over the whole range of the m/z spectra. This survey scan is used to identify the most intense ions in the sample at this moment, for our exemple we will use top 40 ions. The machine next isolates (NB : MS uses a very small window of 0.7 Da around the desired ion to "isolate" the precursors) and fragments every single one of those top ions individually . The result of this operation is a known precursor ion with every single fragment ions that are associated with this precursor (ex: Precursor : 428.3 m/z. Fragments ions : 324.5, 124.4, 225.9 m/z). Once this is done, the machine starts another cycle. What explains the sequencing depth of the IDA mode is the fact that we can add a restriction to the machine : an ion cannot be fragmented more than 2 times for a selected time in a single run. So once the machine has identified an ion as one of the top 40 most intense ions in the sample in two different cycles, this ion is excluded from the list and the next 40 most intense ions get included in the list and fragmented. And so on.
Question 2 : I don’t understand why the ion library is not generated with SWATH, or why the database is not generated by sequencing every ion in the sample at the MS and MS/MS level
Answer : Ideally, that’s what we should do. In a perfect world, the machine would be fast enough to scan the whole m/z range in small 0.7 Da windows, which correspond to a MRM window and is the smallest window that we can isolate with quadrupoles. Scanning the usual m/z range for proteins (from 350 to 1250 m/z) represents roughly 1285 individual 0.7 Da window in which we have to isolate and fragment the precursor ion to record its fragments ions. However, the actual MS technology is far from this number. With the latest version of our machine, the TripleTOF 6600, one can define 200 larger m/z windows in 1 cycle. With our machine, we can only define 100 windows in 1 cycle. Now you could ask why can’t we generate the database with only 100 larger windows, since we have both the MS and the MS/MS information for every single one of these windows. The answer is that what we get from the analysis of one SWATH window is a mix of all the MS/MS fragments of all the MS ions that were present in this window. Some algorithms are able to deconvolute this mis of MS/MS signal to associate the MS/MS fragments to only one MS ion. However, these algorithms are not perfect. So, if need be, we can generate a database without an IDA acquisition. The way to go is to do a SWATH acquisition and to run a deconvolution algorithm on the data. The result is a list of MS ions plus their deconvoluted MS/MS fragments, which is the exact same thing as the IDA mode.
Question 3 : Are the ions associated to a protein by mapping the results on a theoretical database made from reference sequences? Is the mapping done by the MS/MS identification software?
Answer : This is exactly what happens. Once the IDA run is completed, the MS and MS/MS information of every ion is mapped on a theoretical database to identify peptides and proteins present in the samples. This process can be done by several algorithm including Mascot, OMSSA, ProteinPilot or multi algorithm interface such as search GUI.
Question 4 : You specify that the SWATH acquisition acquires the data by analyzing the windows sequentially. Normally, how is the data acquired?
Answer : Normally, or in IDA mode, the data is acquired after the survey scan. The survey scan identifies the 40 most intense ions and only fragments these ions. This means that at a fixed time points, the only ions that are fragmented are the selected most intense one. In SWATH, in 1 cycle, all the windows are acquired sequencially. Ex : Let’s say we want to cover a range of ions from 350 to 1250 m/z. The window #1 could range from 350 to 352.4 m/z. The machine will fragment all the ions (intense or not) that fit in this window. The result will be a mix of MS/MS fragments associated with all the precusors within the 350-352.4 m/z window. Then, the machine will isolate and fragment the ions from the second window, let’s say from 352.4 to 356.1 m/z to record their MS/MS fragments, and so on, until the 100th window. It is pretty hard to describe, but SCIEX has made a pretty good video on the matter.
Differences between cycles in IDA vs SWATH
Question 5 : You did not talk about the cons of the SWATH acquisition. Are there any?
Provided you already have the ion library, a SWATH analysis is not really more expensive than a shotgun analysis. The data analysis is more complicated on our end, but that does not affect the end user. If you don’t have an ion library (and do not want to generate one by IDA, which would cost more), we can run our algorithms on the SWATH data and generate one for a little extra. A SWATH analysis does not require more starting material, more sample prep and does not take more machine time than a shotgun proteomic analysis. So there are almost no reasons to choose IDA over SWATH for your proteomics, metabolomics or lipidomics experiments!

Conclusions
We hope these answers clarified some of the interrogations you had on SWATH. Don't hesitate to write back to us if there are still somewhat confusing points that you would like to clarify.