Next generation SWATH proteomics - more speed and more depth
For most academic researchers or pharma people, mass spectrometry is just a way to quantify a molecule of interest or to identify what is hidden in that silver-stained band. The reality is that the technological advances in mass spectrometry made that machine much more powerful. Sure, one can still quantify a given product very accurately, and at tiny concentrations using MRM acquisition. But what if someone wanted to analyze 1000s variations in the protein content (proteome) of a cell line between two treatments? What if they wanted to correlate RNA sequencing data with the actual protein levels? What if they wanted to look, identify and quantify clinically relevant biomarkers in cancerous tissues? Well, that’s what the MS/MSall technology from SCIEX, termed SWATH is for.
So, what about SWATH?
Sorry that one was too easy! When reading about mass spectrometry, you often see the terms “MS” and “MS/MS”. “MS” (or MS1) level of analysis means that the machine measures the intact molecule (parent molecule), while the “MS/MS” (or MS2) level refers to the analysis of the fragments (product ions) that make the intact molecule. This analysis requires the machine to isolate the intact molecule and to break it, using electrical current and gas to produce specific signature ions from the parent molecule. These ions can then be used to identify a molecule and to quantify it.
This is all nice, but why should we care about SWATH acquisition? Shotgun proteomics has been used for more than 20 years to study proteome variation! Simple fact: standard shotgun proteomics uses data dependent acquisition, while a SWATH analysis is data independent (DIA) meaning it sequentially records the MS/MS of every molecule in the samples (some of you may have noticed the reference to MS/MSall in the blog title). This innovation was published by a group working with Ruedi Aebersold and termed it SWATH in reference to the swaths; a series of acquired isolation windows.
Why is SWATH acquisition so wonderful?
While DDA/IDA have strong computer- and sample-based bias, SWATH brings a new level of reproducibility and quantification depth in proteomics by removing any data based bias (SCIEX has made a very good explanatory video on the subject). This brings to two major implications:
- Since the odds of two different molecules having the same MS and MS/MS signature at high resolution is very low, the chances of getting interfering compounds are very low.
- The way the method is developed means that there is no need to know beforehand what you will be quantifying.
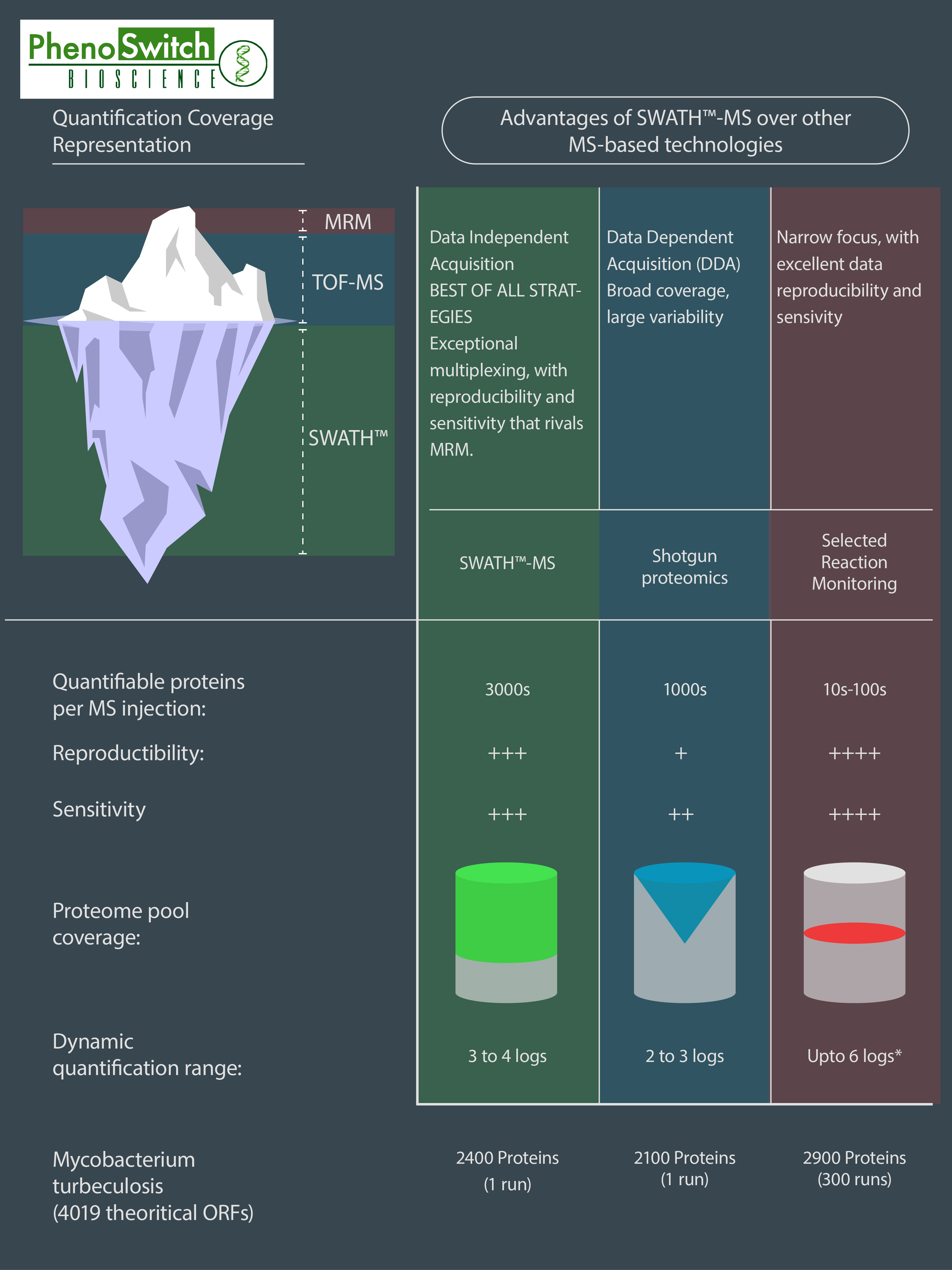
If we do know what we want to quantify, we can use another interesting feature of the SWATH technology : the customizable windows size. This means that we can narrow some of the windows to fit to one or several known molecules we are particularly interested in, while still acquiring data for everything else! That way, we can get almost as precise in a SWATH analysis as we are in MRM, which is our most precise and sensitive acquisition mode. To better understand the differences between SWATH proteomics, shotgun proteomics (normally used for global protein identification) and MRM, please refer to the following infographics.
.png?width=759&height=1012&name=SWATH%20Acquisition%20(2).png)
This infographics was inspired by data obtained in several research articles (read the full description of the figure in the application note)
How can you quantify something that you don’t know is there?
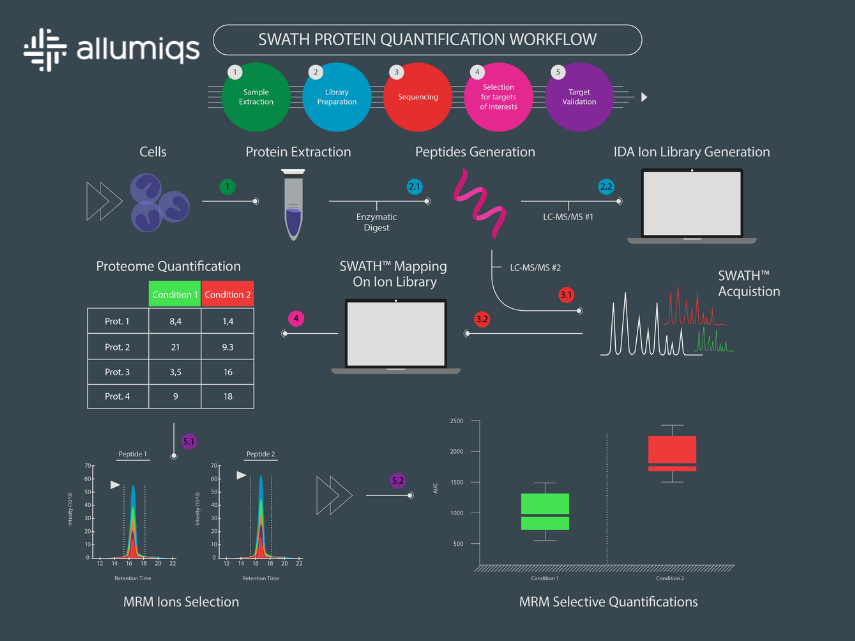
The answer to this question is in the ion library that we use (see the application note for more details). Once the acquisition is done, the files are processed by powerful bioinformatics tools that use the ion library to deconvolute the signal and get clean precursor MS and their MS/MS information. These data can then be mapped on peptide, lipid or metabolite databases to generate quantitative information. With this method, we have been able to get reproducible quantitative data for more than 4000 proteins. That’s pretty impressive! Also, since the acquisition is data independent, the files can be mapped over and over again on upgraded, changed or tweaked databases as new hypotheses rise. You can thus squeeze even more information out of the data files.
Why do they call it Next Generation Proteomics (NGP)?
Simple: it resembles a lot to next generation sequencing (NGS)! To illustrate the general workflow of a SWATH proteomics experiment, we have designed an infographic comparison between an RNAseq and a SWATH experiment (read the full description of the figure in the application note).


What do I need to benefit from SWATH proteomics?
Not much, really! Allumiqs offers complete solutions for all your projects. You provide the biological material (pelleted cells, frozen tissues, plasma, etc) and we do the lysis/extraction, digestion, purification and LC-MS/MS analysis. Once the samples have been processed, we generate a complete data report that you can easily understand and benefit from. We provide custom made reports in the form of a spreadsheet, in a format that you can easily understand. If need be, we can arrange meetings to answer all your questions or clarify certain points on the data report/method we used. We even write your material and methods!

After the publication of this blog post, we received a few questions about the SWATH technology from our readers. To clarify our explications and in the hopes of generating useful content, we wrote another blog post in the form of a Q&A about SWATH acquisition. Hope you enjoy!